
Etai Jacob, PhD
Computational Biologist
Dana-Farber Cancer Institute | ejacob [AT] jimmy.harvard.edu
Broad Institute of MIT and Harvard | ejacob [AT] broadinstitute.org
Lattice model protein folding simulations
Why are terminal residues of proteins exposed?
Studying the fundamental questions underlying the phenomenon of protein folding has been facilitated by the introduction of simple folding models simulations. Simple, or even abstract, models of protein folding, while ignoring many of the small details of this process, are very useful for elucidating general principles regarding protein folding. Clearly, simple model experiments can not be used to definitively prove specific folding phenomena, but conclusions from simple models can certainly be used to promote and critically assess ideas about protein folding mechanisms. In this research I used one of the major simple folding models - the lattice model. In this model, the polypeptide chains in the simulations are modeled as a linear sequence of residues on a 2D lattice, where a reduced number of residues types are used and interaction potentials between residues reflect the average strength of interactions in empirical mean force potentials.
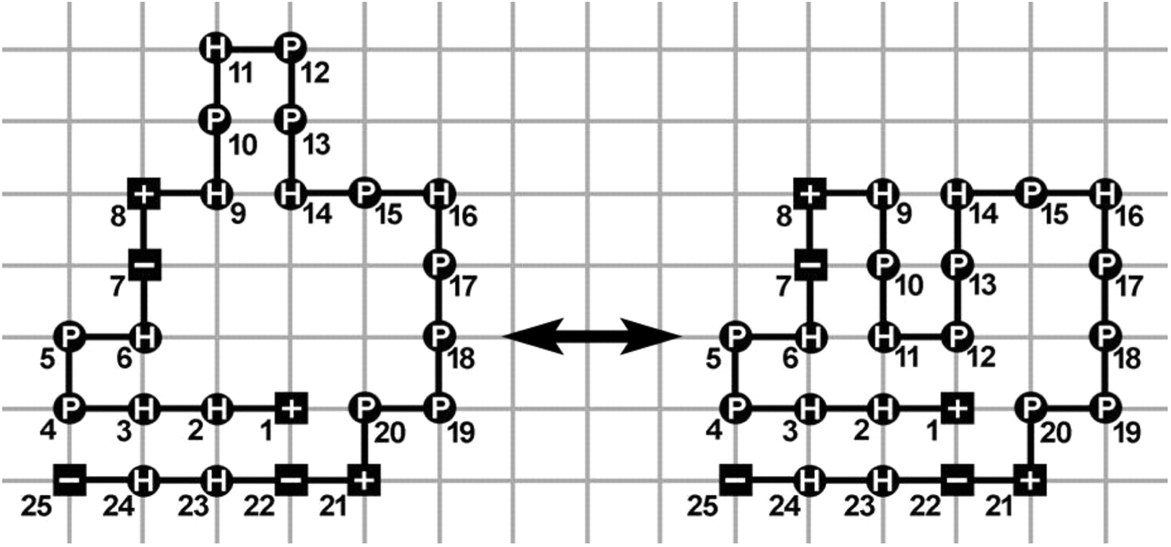
An example of a protein lattice model.
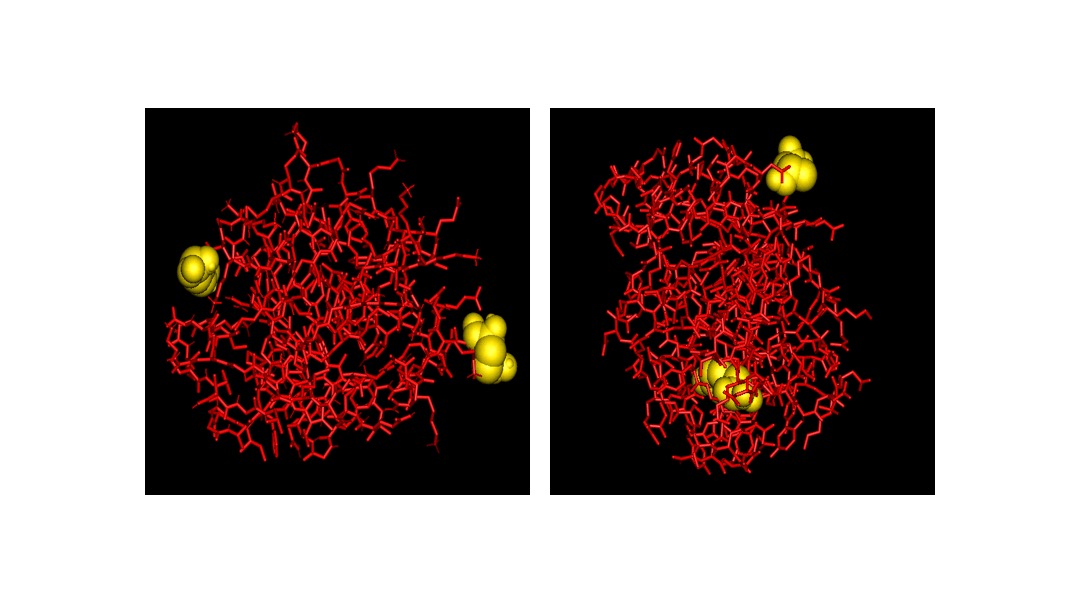
An example of terminal residues of real proteins. For most proteins both termini are exposed to the solvent, as in cytochrome c552 (PDB code 1C52) (left, where terminal residues are shown as yellow space filling objects). Only in very few cases, termini residues are buried as in (right) staphopain (code 1CV8) a cysteine proteinase where the C-terminal tyrosine is totally buried.
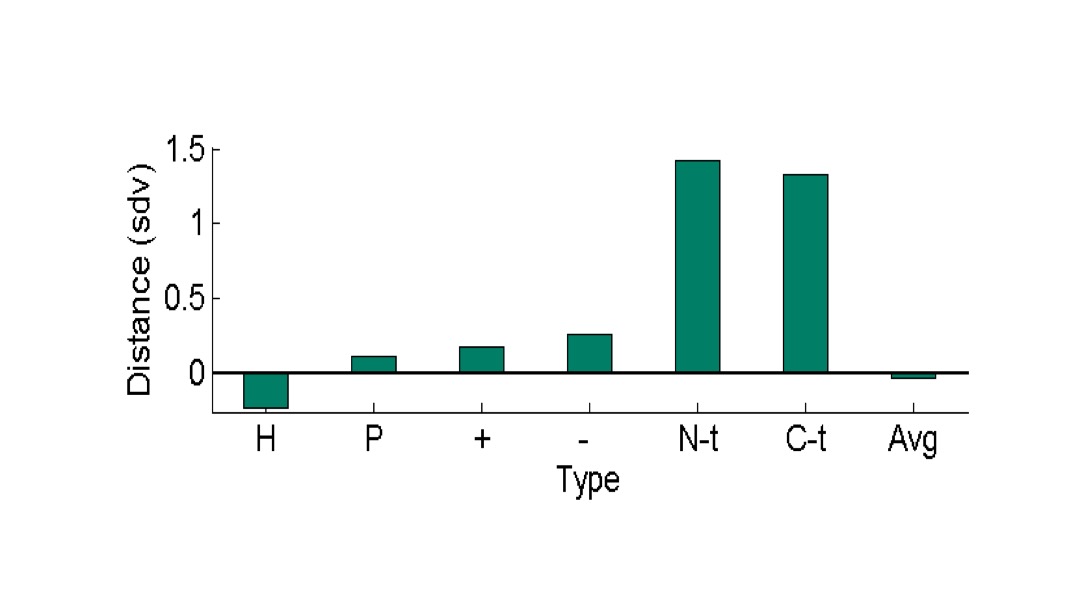
The distance of residues from the center of PDB proteins.
For the dataset of 425 proteins, the distance of residues to the center of mass of their proteins is presented. The average distances, in units of standard deviations of distances in each protein, are grouped by residue type. It is evident that terminal residues are most distant from the center of their proteins.
This work was published in Bioinformatics
Source code
The source code is included in a C++ software suite:
- A Software suite to perform enumeration and simulations of a simplified model (i.e. toy model) of protein structure and folding process: TOYFOLD.