
Etai Jacob, PhD
Computational Biologist
Dana-Farber Cancer Institute | ejacob [AT] jimmy.harvard.edu
Broad Institute of MIT and Harvard | ejacob [AT] broadinstitute.org
Protein-chaperonin interactions
Computational studies of protein chaperone mediated folding interactions
In this work I studied the folding process of proteins with the aid of two models of molecular chaperones; prokaryotic and eukaryotic. One major class of chaperones, called chaperonins, comprises ATP-dependent proteins that facilitate folding by binding the assisted protein in a cavity formed at each end of their double ring structure. The prokaryotic GroEL/GroES complex in E. coli is the best characterized chaperonin complex. GroEL consists of two rings each formed by seven identical protein subunits. GroES is a single-ring heptamer that binds to GroEL in the presence of ATP and serves as the cap of the cavity formed by each ring structure. Each GroEL subunit can rotate and thus turn a different surface towards the inner cavity. The homologue eukaryotic chaperonin to the prokaryotic chaperonin GroEL is called CCT (also called TCP-1 ring complex) which is composed of eight similar (but not identical) subunits.
One major difference between the prokaryotic and eukaryotic chaperonins is the coordination between the surface changes of the subunits. While in the GroEL/GroES system the change is concerted, i.e. all subunits switch simultaneously, it was shown that in CCT the change is sequential, i.e. the subunits switch conformation one after the other. Concerning the substrates, approximately 70% of proteins in eukaryotic cells are multi-domain whereas in prokaryotes single-domain proteins are more common. Thus, it was suggested that the different modes of action of prokaryotic and eukaryotic chaperonins can be explained by the need of eukaryotic chaperones to facilitate folding of multi-domain proteins. In this study, I examined possible implications of these different allosteric mechanisms for the assistance ability of these chaperonins to folding process of proteins.
In order to enable the simulations of the protein folding process and the protein chaperon interactions in a simple and abstract model, I have developed a computational engine that can be used to simulate many types of interactions between a folding protein and a chaperone on a lattice. Using this engine, it is possible to specify many parameters of the system including the chaperonin’s shape, size, surface composition, the way the chaperonin changes its surface, the strength of interactions between amino acids etc. In addition, this engine can generate different substrates; single-domain proteins, double-domain proteins, with different length and with various kinetic, structural or content features.
Using this software simulation engine, I found that the folding yields of single-domain protein substrates are greater when the chaperonin undergoes concerted and not sequential conformational changes. In contrast, the folding yields of double-domain proteins are greater in the presence of a chaperonin that undergoes sequential conformational changes. These results are consistent with the observation that large multi-domains proteins are more common in eukaryotes compared with prokaryotes. Hence, they support the suggestion that the different allosteric mechanisms of prokaryotic and eukaryotic chaperones can be explained by the need of eukaryotic chaperones to facilitate folding of proteins with a multi-domain structure.
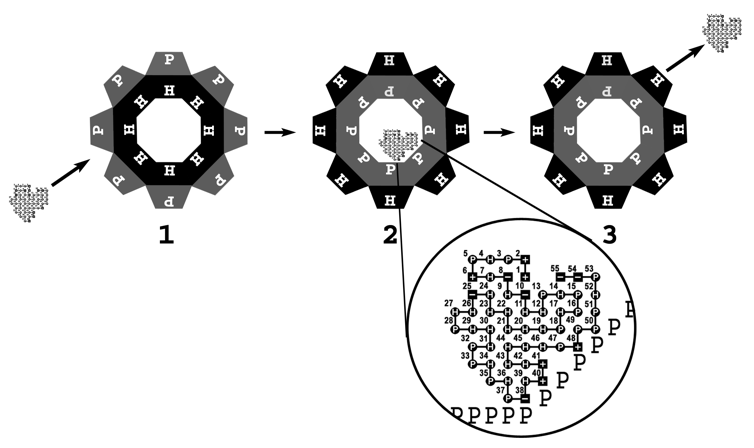
Schematic view of a chaperonin that undergoes a concerted surface change.
In this figure, an octameric chaperonin is depicted. (1) The chaperonin binds a protein substrate in its hydrophobic cavity. (2) The cavity surface switches from fully hydrophobic to fully polar. (3) After a predefined period that a protein substrate spends inside the chaperonin cavity, it is ejected outside. The magnified region shows a 55 residuesingle-domain protein interacting with a polar surface of a chaperonin cavity. Three charged-polar interactions are present between the surfaces of the chaperonin and the protein substrate. This concerted mode of surface change characterizes the GroEL/S prokaryotic chaperonin.
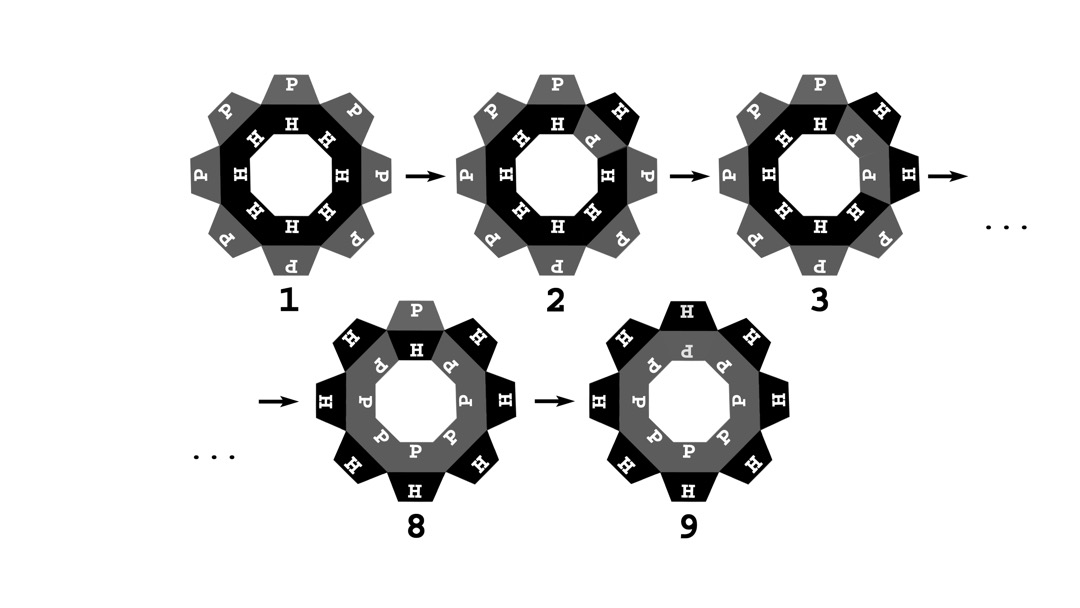
A schematic view of a chaperonin that undergoes a sequential surface change. In this figure, an octameric chaperonin is depicted. (1) The chaperonin cavity in the substrate protein-bound state is initially fully hydrophobic. (2) The first change occurs when one side of the octagonal structure “flips” to a polar conformation. (3) In the subsequent stages, the adjacent subunits also switch to a polar conformation. (8,9) The process of sequential alteration of the cavity sides from hydrophobic to polar ends when all eight of them are polar. The time between each additional change is predefined for each simulation and is constant during the whole simulation. This model represents the eukaryotic chaperonin (CCT).
This work was published in Bioinformatics
Source code
The source code is included in a C++ software suite:
- A Software suite to perform enumeration and simulations of a simplified model (i.e. toy model) of protein structure and folding process: TOYFOLD.