
Etai Jacob, PhD
Computational Biologist
Dana-Farber Cancer Institute | ejacob [AT] jimmy.harvard.edu
Broad Institute of MIT and Harvard | ejacob [AT] broadinstitute.org
Correlated mutation analysis
Codon-level information improves predictions of inter-residue contacts in proteins
Abstract. Genomic sequences contain rich evolutionary information about functional and structural constraints on proteins. This information can be mined to detect correlated mutations in proteins and address the long-standing challenge of predicting protein three-dimensional structures from amino acid sequences. Methods for analysing correlated mutations in proteins are becoming an increasingly powerful tool for predicting contacts within and between proteins owing to the explosive growth in sequence data and significant theoretical progress. Nevertheless, limitations remain due to the requirement for large multiple sequence alignments (MSA) and the fact that, in general, only the relatively small number of top-ranking predictions are reliable. Previously, methods for analysing correlated mutations have relied exclusively on amino acid MSAs as inputs. In this work, I describe a new approach for analysing correlated mutations that is based on combined analysis of amino acid and codon MSAs. I show that a direct contact is more likely to be present when the correlation between the positions is strong at the amino acid level but weak at the codon level. The performance of different methods for analysing correlated mutations in predicting contacts is shown to be enhanced significantly when amino acid and codon data are combined.
This work was published in eLife

Example of a pairwise correlation in a multiple amino acid sequence alignment and two possible corresponding codon alignments.
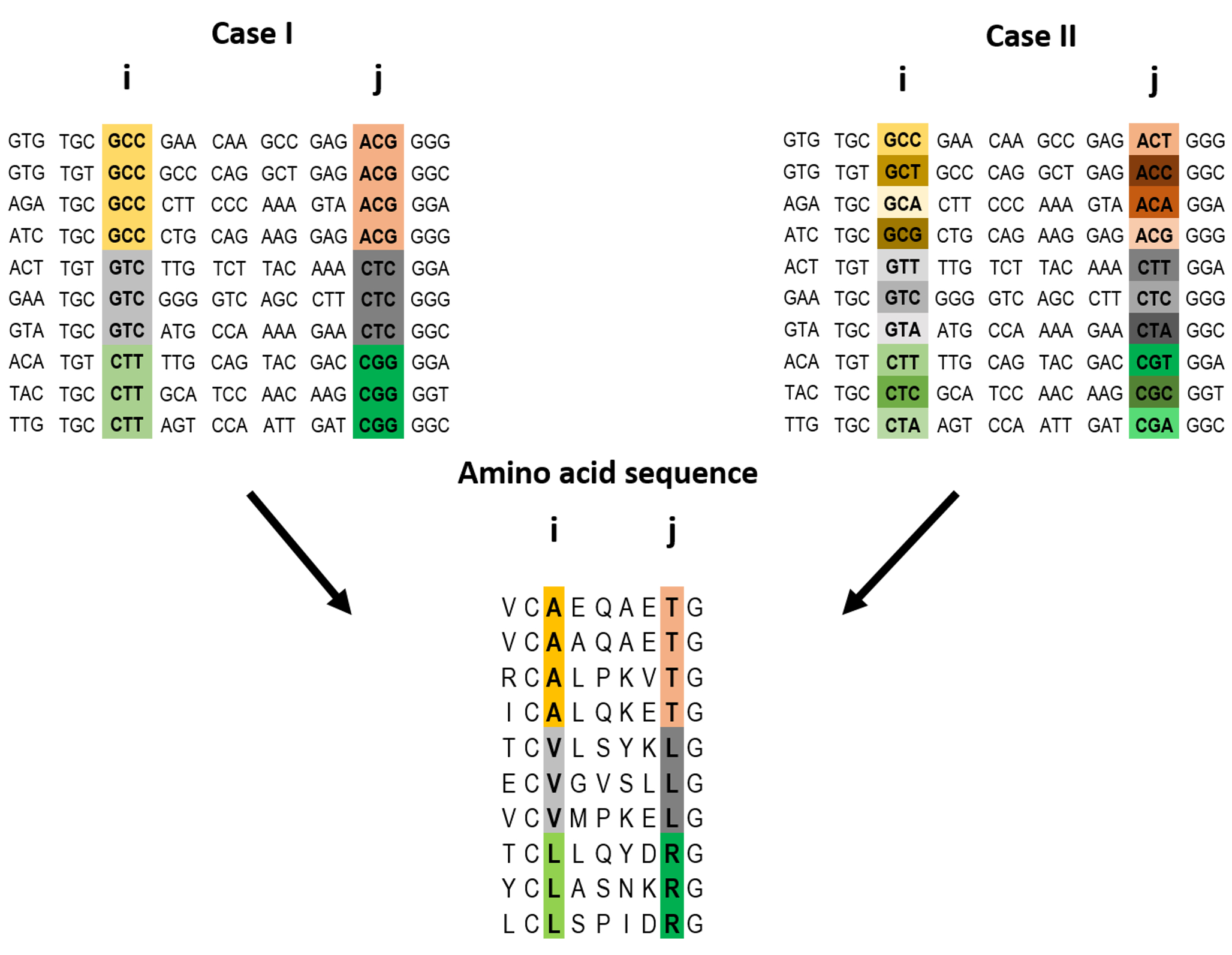
A correlation at the amino acid level between two positions i and j may (top left) or may not (top right) be accompanied by a correlation at the codon level. The premise of the method introduced here is that a correlation at the amino acid level between two positions is more likely to reflect a direct interaction if the correlation at the codon level for these positions is weak (top right).
Source code
The source code is included in four different repositories:
- An R package to compute correlated mutations using both codon and amino acid levels: CMA.
- An R package for mapping Pfam multiple sequence alignment to PDB residue coordinates: MAPDB.
- A python/C++ program to compute protein Sparse Inverse COVariance analysis program for nucleotide and amino acid sequences: ACPSICOV.
- Python code for generating codon MSA file for a given amino acid MSA file: AA2CODON.